N-Heteropolyzyklen als Funktionsmaterialien
B07 - Theoretical Modeling of Small Clusters of N-heteropolycycles
Subject Area General Theoretical Chemistry
Dr. Höfener – PCI, KIT
Project Description
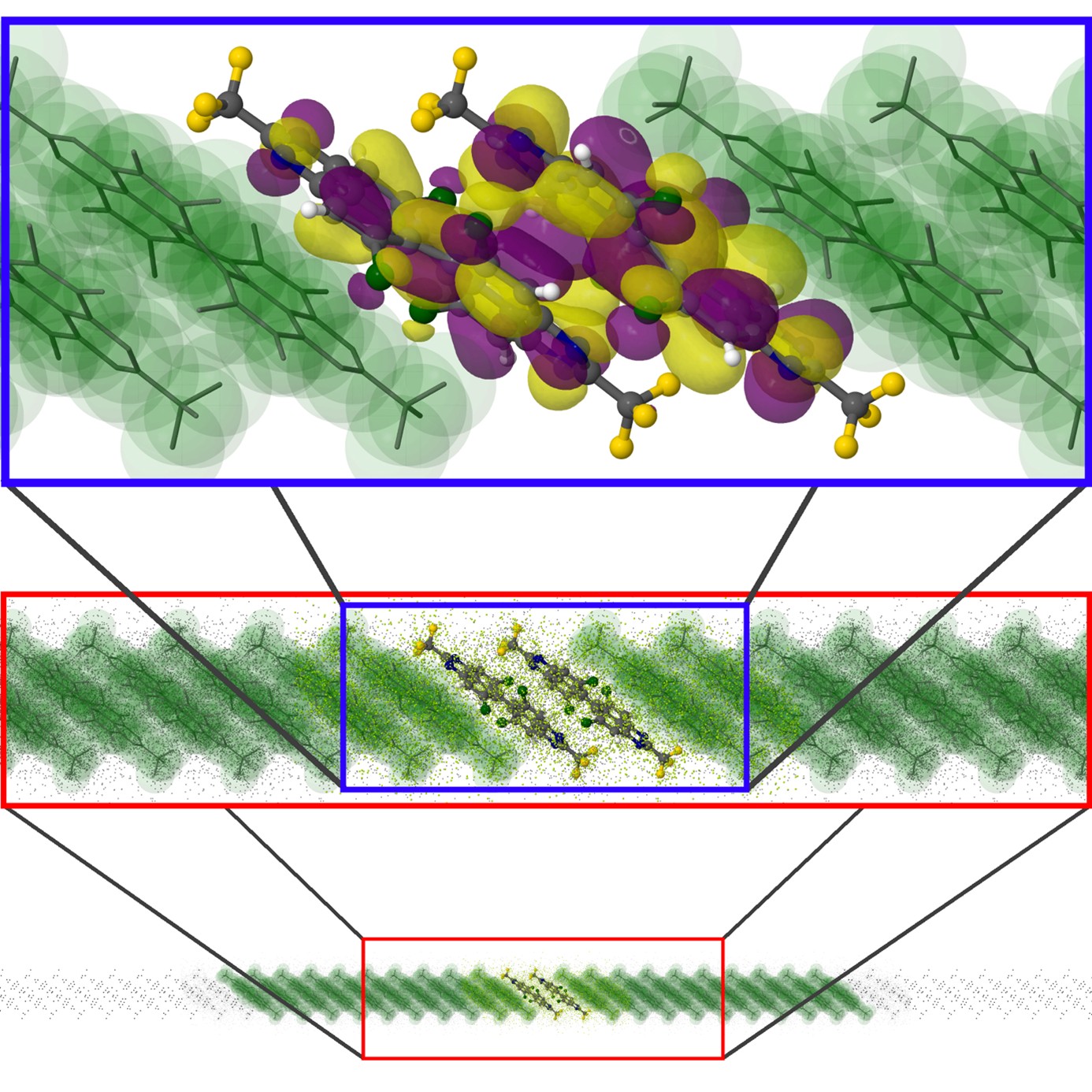
Molecular vibrations are crucial for charge transport in van der Waals molecular crystals of organic semiconductors. The identification of specific modes contributing to electron-phonon coupling is therefore crucial for understanding the charge transport properties of an organic semiconductor. In this subproject, new specialized methods will be developed using frozen-density embedding (FDE) to describe electron-phonon couplings with the necessary accuracy while ensuring applicability to molecular assemblies. This combination ensures that intermolecular vibrations can be calculated in realistic dimers, since true local minima are present due to the crystal. The newly developed quantum chemical methods represent a suitable approach to gain an understanding of intermolecular vibrations in crystal environments. Of crucial importance is furthermore the knowledge of the overlap of the ionized electron densities, which are on the one hand repulsive and on the other hand explicitly describe the charge transport by transferring the charge. The developed methods will be investigated in close collaboration with experimental groups for N-heteropolycycles by measuring relevant low-lying Raman modes in organic semiconductors, for example tetraazaperopyrenes (TAPPs).